こんにちは!ぼりたそです!
今回はPythonのライブラリであるRDkitを使って化合物の構造最適化を実行する手法について解説していきたいと思います。
この記事は以下のポイントでまとめています。
- 構造最適化の実行コード
- 化合物の3D構造生成
- 力場の作成(UFF, MMFF)
- 構造最適化
それでは順番に説明していきます。
構造最適化の実行コード
それでは、まず構造最適化の実行コードをご紹介します。
また、このコードは化合物の描画も含んでいるため、jupyter notebook上での実行を前提としています。
import rdkit
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.rdForceFieldHelpers import MMFFGetMoleculeForceField
import py3Dmol
import matplotlib.pyplot as plt
def optimize_molecule(smiles, force_field_type='UFF', max_steps=1000, force_tolerance=1e-3):
"""
分子の構造最適化を行い、エネルギーの変化をプロットする関数。
Args:
mol (Mol): RDKitの分子オブジェクト。
force_field_type (str): 使用する力場の種類('UFF', 'MMFF')。デフォルトは 'UFF'。
max_steps (int): 最適化の最大ステップ数。デフォルトは1000。
force_tolerance (float): 収束条件としての勾配の閾値。デフォルトは1e-3。
"""
# 分子を読み込む
mol = Chem.AddHs(Chem.MolFromSmiles(smiles))
# 3D座標を生成する
AllChem.EmbedMolecule(mol, randomSeed=42)
if force_field_type == 'UFF':
# 力場の作成(デフォルトはUFFを使用)
force_field = AllChem.UFFGetMoleculeForceField(mol)
elif force_field_type == 'MMFF':
# MMFF用のプロパティを取得
props = AllChem.MMFFGetMoleculeProperties(mol)
# MMFF力場を作成
force_field = MMFFGetMoleculeForceField(mol, props)
# エネルギーの変化を記録するリスト
energies = []
# 構造最適化を実行し、エネルギーを記録
for step in range(max_steps):
# 最適化の1ステップ実行
converged = force_field.Minimize(maxIts=1, forceTol=force_tolerance)
# 現在のエネルギーを記録
energy = force_field.CalcEnergy()
energies.append(energy)
print(f"Step {step}: Energy = {energy:.6f} kcal/mol")
# 勾配(力)の確認
max_force = max(force_field.CalcGrad())
print(f"Maximum Force: {max_force:.6f} kcal/mol/Å")
# 勾配が閾値以下であれば終了
if max_force < force_tolerance:
print("Optimization converged.")
break
# 最適化後のエネルギー
final_energy = force_field.CalcEnergy()
print(f"Final Energy: {final_energy:.6f} kcal/mol")
# エネルギーの推移をプロット
plt.figure(figsize=(8, 6))
plt.plot(energies, marker='o', linestyle='-', color='b')
plt.title(f'Energy Convergence During {force_field_type} Optimization')
plt.xlabel('Optimization Step')
plt.ylabel('Energy (kcal/mol)')
plt.grid(True)
plt.show()
# 分子をスティックスタイルで表示
mb = Chem.MolToMolBlock(mol)
viewer = py3Dmol.view(width=400, height=400)
viewer.addModel(mb, 'mol')
viewer.setStyle({'stick': {}})
viewer.zoomTo()
viewer.show()
#計算する化合物のSMILESを定義
smiles = 'C1CCCCC1C'
# 最適化の実行(MMFF力場使用)
optimize_molecule(smiles=smiles, force_field_type='MMFF')
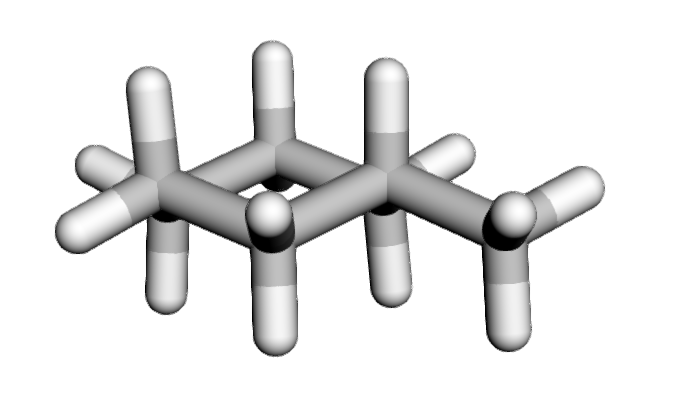
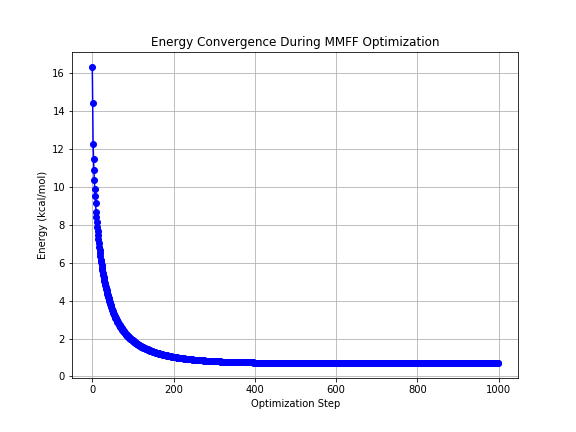
実行すると以下のように構造最適化が終了した構造とエネルギーの収束のプロットが出力されます。
今回は例としてメチルシクロヘキサンの構造最適化を実行しましたが、きちんと椅子型の構造をとっており、かつメチル基が安定なエカトリアル位の配座を取っています。ちなみにpy3Dmolで描画した分子はスクロールやクリックなどで分子の回転や拡大縮小が自由自在にできます。
また、エネルギーの推移を見てもほぼ収束していることがわかります。
このコードは以下の大きく3つのプロセスから処理されています。
- 化合物の3D構造の生成
- 力場の作成
- 構造最適化
以下、詳細に説明していきます。
化合物の3D構造の生成
まずは化合物の3D構造の生成について説明していきます。
実行コードの中では以下の部分で処理しています。
# 分子を読み込む
mol = Chem.AddHs(Chem.MolFromSmiles(smiles))
# 3D座標を生成する
AllChem.EmbedMolecule(mol, randomSeed=42)各メソッドについては以下の通りの説明となります。
Chem.MolFromSmiles(smiles):
- SMILESからRDKitの分子オブジェクトを生成しています。今回であれば、 “C1CCCCC1C” (メチルシクロヘキサン)の構造を表します。
Chem.AddHs(mol):
- 3D構造生成には水素が必要ですが、SMILESは通常水素を明示しないため、生成した分子に水素原子を付与しています。
AllChem.EmbedMolecule(mol, randomSeed=42):
- 分子に3D構造を付与しています。内部では、分子の結合情報を基にして、エネルギー的に妥当な3D構造を生成します。
randomSeed=42により、毎回同じ3D構造が生成されるようになっています。
力場の作成
次は力場の作成について説明していきます。
実行コードの中では以下の部分で処理しています。
if force_field_type == 'UFF':
# 力場の作成(デフォルトはUFFを使用)
force_field = AllChem.UFFGetMoleculeForceField(mol)
elif force_field_type == 'MMFF':
# MMFF用のプロパティを取得
props = AllChem.MMFFGetMoleculeProperties(mol)
# MMFF力場を作成
force_field = MMFFGetMoleculeForceField(mol, props)
力場については「UFF(Universal Force Field)」と「MMFF(Merck Molecular Force Field)」のどちらかを選択できるようにしています。また、各メソッドについては以下のような役割を果たしています。
UFFGetMoleculeForceField(mol):
- RDkitの分子オブジェクト(mol)を入力することで分子に対してUFF力場を適用します。
MMFFGetMoleculeForceField(mol, props):
MMFFGetMoleculeProperties(mol)で、官能基や結合情報などMMFF力場に必要なパラメータをpropsとして分子から取得します。これに基づき、RDkitの分子オブジェクトからMMFF力場を生成します。
ちなみに「UFF」と「MMFF」は以下のような違いがあります。
| 比較項目 | UFF(Universal Force Field) | MMFF(Merck Molecular Force Field) |
|---|---|---|
| 特徴 | 汎用的。ほとんどの元素に適用可能。 | 有機化合物に特化。化学的特性に基づいた経験的パラメータを使用 |
| 精度 | 汎用的なためやや精度は低い | 有機化合物において高精度 |
| 計算速度 | 比較的高速 | UFFに比べると遅い |
| エネルギー項 | 基本的な結合、角度、非結合相互作用など | 基本的な結合、角度、非結合相互作用に加え、より詳細な水素結合や静電相互作用も考慮 |
| 用途 | 広範囲の元素に対しておおまかな計算がしたい場合 | 有機分子の正確な構造最適化をしたい場合 |
構造最適化
最後に構造最適化について説明します。
構造最適化は実行コードの以下の部分で処理しています。
# エネルギーの変化を記録するリスト
energies = []
# 構造最適化を実行し、エネルギーを記録
for step in range(max_steps):
# 最適化の1ステップ実行
converged = force_field.Minimize(maxIts=1, forceTol=force_tolerance)
# 現在のエネルギーを記録
energy = force_field.CalcEnergy()
energies.append(energy)
print(f"Step {step}: Energy = {energy:.6f} kcal/mol")
# 勾配(力)の確認
max_force = max(force_field.CalcGrad())
print(f"Maximum Force: {max_force:.6f} kcal/mol/Å")
# 勾配が閾値以下であれば終了
if max_force < force_tolerance:
print("Optimization converged.")
break
# 最適化後のエネルギー
final_energy = force_field.CalcEnergy()
print(f"Final Energy: {final_energy:.6f} kcal/mol")
# エネルギーの推移をプロット
plt.figure(figsize=(8, 6))
plt.plot(energies, marker='o', linestyle='-', color='b')
plt.title(f'Energy Convergence During {force_field_type} Optimization')
plt.xlabel('Optimization Step')
plt.ylabel('Energy (kcal/mol)')
plt.grid(True)
plt.show()
# 分子をスティックスタイルで表示
mb = Chem.MolToMolBlock(mol)
viewer = py3Dmol.view(width=400, height=400)
viewer.addModel(mb, 'mol')
viewer.setStyle({'stick': {}})
viewer.zoomTo()
viewer.show()このコードは構造最適化を実行し、構造のエネルギー準位の推移をプロットしつつ、最終的な構造を3Dで表示できるようになっています。
構造最適化の実行においては以下メソッドの説明を記載いたします。
force_field.Minimize(maxIts=1, forceTol=force_tolerance):
- 1ステップだけ最適化(エネルギー最小化)を行います。エネルギーを最小化するために、各原子にかかる力(勾配)を減少させる動作を行います。
maxIts=1は1ステップずつ最適化を実行することを指定しています。これにより、エネルギーや勾配の変化を細かく確認できます。
energy = force_field.CalcEnergy():
- 現在の構造におけるエネルギーを計算します。これがポテンシャルエネルギー(分子内の結合や相互作用によるエネルギー)です。
max_force = max(force_field.CalcGrad()):
- 各原子にかかる力(勾配)の最大値を取得します。この力が設定した収束条件(
force_tolerance)より小さければ、分子が安定した状態に近づいていると判断しています。
最終的にはエネルギー勾配が閾値以下(エネルギーが収束)になるか既定した最適化の回数を満たすと終了するようになっています。
また、py3Dmolで3D分子を描画においては以下メソッドの説明を記載いたします。
Chem.MolToMolBlock(mol):
- 分子の3D構造をMolBlock形式に変換し、
py3Dmolライブラリを使って分子を3D表示します。
viewer.setStyle({'stick': {}}):
- スティックモデル(分子の結合を棒状で示す形式)を指定しています。
zoomTo():
- 分子全体が画面に収まるようにズーム or ズームアウトします。
終わりに
以上がRDkitを使用した化合物の構造最適化についての説明になります。UFFやMMFFによる構造最適化は量子化学計算と比較すると精度は劣るかもしれませんが、ざっくり計算したい場合などには使える気がします。また、分子MDの計算などにはこの手の力場を使用して計算するケースもあるかと思います。この記事が皆さんの一助になることを願っております。

