こんにちは!ぼりたそです!今日はPythonで化合構造を記述子に変換する方法について解説していきます。
この記事は以下のポイントでまとめています。
- 記述子とは?
- Mordred記述子について
- PythonでMordred記述子を出力
記述子とは後程ご説明しますが、簡単に言えば化合物の特徴を数値化したデータになります。主に化合物を使用した機械学習などで使用されるものであり、今回はPythonで記述子を出力したいと思います!
記述子とは?
冒頭にも登場しましたが、記述子とはなんだろうと思った方も多いかと思います。某も大学院まで化学を専攻していましたが、分野が違うのかMI(マテリアルインフォマティクス)を学ぶまでは聞いたこともありませんでした。
記述子とは化合物の構造や特徴などを数値化するパラメータのことを言います。データサイエンスの分野では化合物の特徴を数値化し、その数値をもとに化合物の反応性や性質などを予測することに利用されます。
例えば
- 分子量
- 炭素の数
- 単結合の数
- 芳香環の数
なども化合物の特徴を表す記述子と言えるでしょう。
Mordred記述子について
今回は記述子の中でもMordred記述子を使用して化学構造を記述子に変換していきたいと思います。
Mordred記述子とはその名の通り記述子の一つであり、平面構造の情報だけで1613種類、立体構造の情報も含めると1826種類の記述子に変換することができます。ここで一つ一つの記述子について説明はできませんが、非常に数が多いので化学構造の情報を正確に表現できているのではないかと考えられます。
また、PythonのライブラリとしてmordredがありPython上でも動かすことができるので便利なんですね。
Mordredの詳細については以下のgithubより参照いただければと思います。
Mordred_github ↓↓↓↓
https://github.com/mordred-descriptor/mordred
PythonでMordred記述子を出力
それでは早速Pythonを使用してMordred記述子を出力していきたいと思います!
その前にもしmordredのライブラリをインストールしていませんでしたら、以下のコマンドでインストールを完了させてください
pip install mordred今回はクロロベンゼン、フェノール、アニリンについてmordred記述子を出力したいと思います。
コードはgithubを参照し以下のように記述しました。
from rdkit import Chem
from mordred import Calculator, descriptors
# 計算機クラスをインスタンス化
calc = Calculator(descriptors, ignore_3D=True)
#SMILESを定義
smiles = ['c1ccccc1Cl', 'c1ccccc1O', 'c1ccccc1N']
# 分子を計算
mols = [Chem.MolFromSmiles(smi) for smi in smiles ]
# pandasでデータフレーム化
df = calc.pandas(mols)
df.index = smiles
df.iloc[:,0:3]
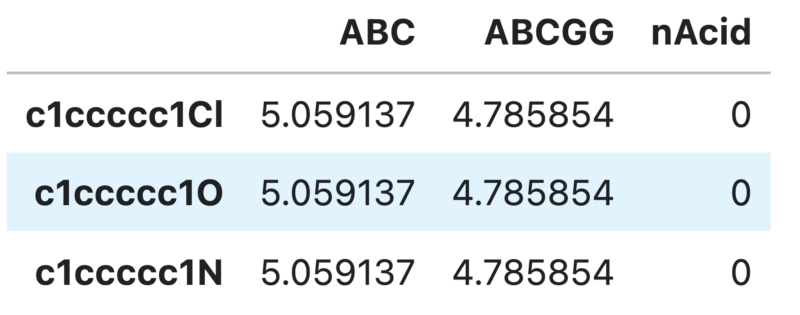
全て表示すると膨大な量になってしまうので最初の3つだけ記述子を表示させました。3つの化合物についてそれぞれ記述子が表示できていますね。
このように記述子をデータフレーム化しておくと機械学習やデータ分析にすぐ利用できるのでとても便利です。
3DのMordred記述子を出力
次に3D情報を含むMordred記述子を出力する方法について紹介します。
3DのMordred記述子を出力するにはCalculatorクラスの引数であるignore_3D=Falseにする必要があります。
しかし、ただignore_3D=Falseにしても適切な計算は行われず、化合物の3次元構造を読み込ませる必要があります。
化合物の3次元構造を取得する方法はいくつかありますが、rdkitを使用してSMILESから3次元構造を計算するのが手っ取り早いです。
実際に3DのMordred記述子を計算するコードは以下の通りです。
from rdkit import Chem
from rdkit.Chem import AllChem
from mordred import Calculator, descriptors
# 分子のSMILES表記
smiles = "CCO"
# SMILESからRDKitの分子オブジェクトを生成
mol = Chem.MolFromSmiles(smiles)
# 3D構造の計算
AllChem.EmbedMolecule(mol, randomSeed=42)
AllChem.MMFFOptimizeMolecule(mol, maxIters=500, nonBondedThresh=200.0)
# Mordredの計算器を作成
calc = Calculator(descriptors, ignore_3D=False)
# 分子記述子の計算
result = calc(mol)
# 結果の表示
print(len(result))
'''
出力結果
1826
'''コードの中にAllChem.EmbedMolecule(mol, randomSeed=42)とありますが、これはmolからSMILESを受け取り初期構造を生成しています。randomSeedによって異なる初期構造が生成されます。
また、AllChem.MMFFOptimizeMolecule(mol, maxIters=500, nonBondedThresh=200.0)とありますが、これは生成した初期構造を最適化するコードになります。
maxItersは最適化の回数でnonBondedThresh=200.0は非結合相互作用のカットオフ半径を設定しています。この距離より遠い非結合原子の相互作用は考慮されないようになります。
このように化合物の3D情報を生成し、Calculatorクラスで計算すると3DのMordred記述子を計算することができます。計算した記述子の数を出力していますが、2次元の時より多く1826個となっています。
終わりに
いかがでしたでしょうか。今回は少しインフォマティクスっぽい記事を書いてみました。化合物のSMILESなどはChemPubなどのオープンソースのデータベースを参照すれば大量に入手できるので、自分で機械学習やデータ分析をやってみたい方にはおすすめです。

