こんにちは!ぼりたそです!今回は化合物の構造式をテキストで表記するSMILES記法について記事をまとめました。
この記事は以下のポイントでまとめています。
- SMILES記法とは?
- SMILESの規則
- SMILESの取得方法
- SMILESの活用例
- オススメの書籍
構造式は視覚的にわかりやすく、化合物が立体的にどのような構造を有しているか一目で理解できるようになっています。
しかし、データベースとして化合物の情報を格納する場合、結合や原子数、不飽和度などの情報は構造式から数値として得られません。
そこで、構造式をテキストで表記できるSMILES記法について今回はご紹介します。
SMILES記法とは?
SMILES(Simplified Molecular Input Line Entry System)は化合物の構造式をテキスト形式で表現するための表記法です。
構造式では人間からすれば視覚的に理解しやすいのですが、機械からしてみられば構造式からは何も理解できないのです。そこで、機械にもわかるように構造式を変換するのがSMILES式なのです。
SMILESは1980年代初頭にDavid Weiningerにより開発されました。Weiningerは化学構造をテキストで表現するための規則的な形式を作り出すことを目的としてSMILESを開発したのです。
開発されたSMILESは化合物の情報処理における標準的な表現方法として広がり、国際化学連合(IUPAC)の機関にでも標準化されています。
その結果、今日では多くの学者や研究者に使われるまでになりました。
SMILESの規則
続きまして、SMILES表記の規則についてご紹介いたします。
今回は以下の規則について説明いたします。
- 元素記号
- 水素原子
- 結合
- 環構造
- 分岐構造
- 幾何異性体
元素記号
化合物の構造中の元素は全てその元素記号で表記されます。例えば炭素であれば「C」、窒素であれば「N」、酸素であれば「O」といった形で表記されます。
例としてブタン、エタノール、エチルアミンの構造式をSMILES表記にしたものを以下に示します。
| 化合物 | 構造式 | SMILES表記 |
|---|---|---|
| ブタン | 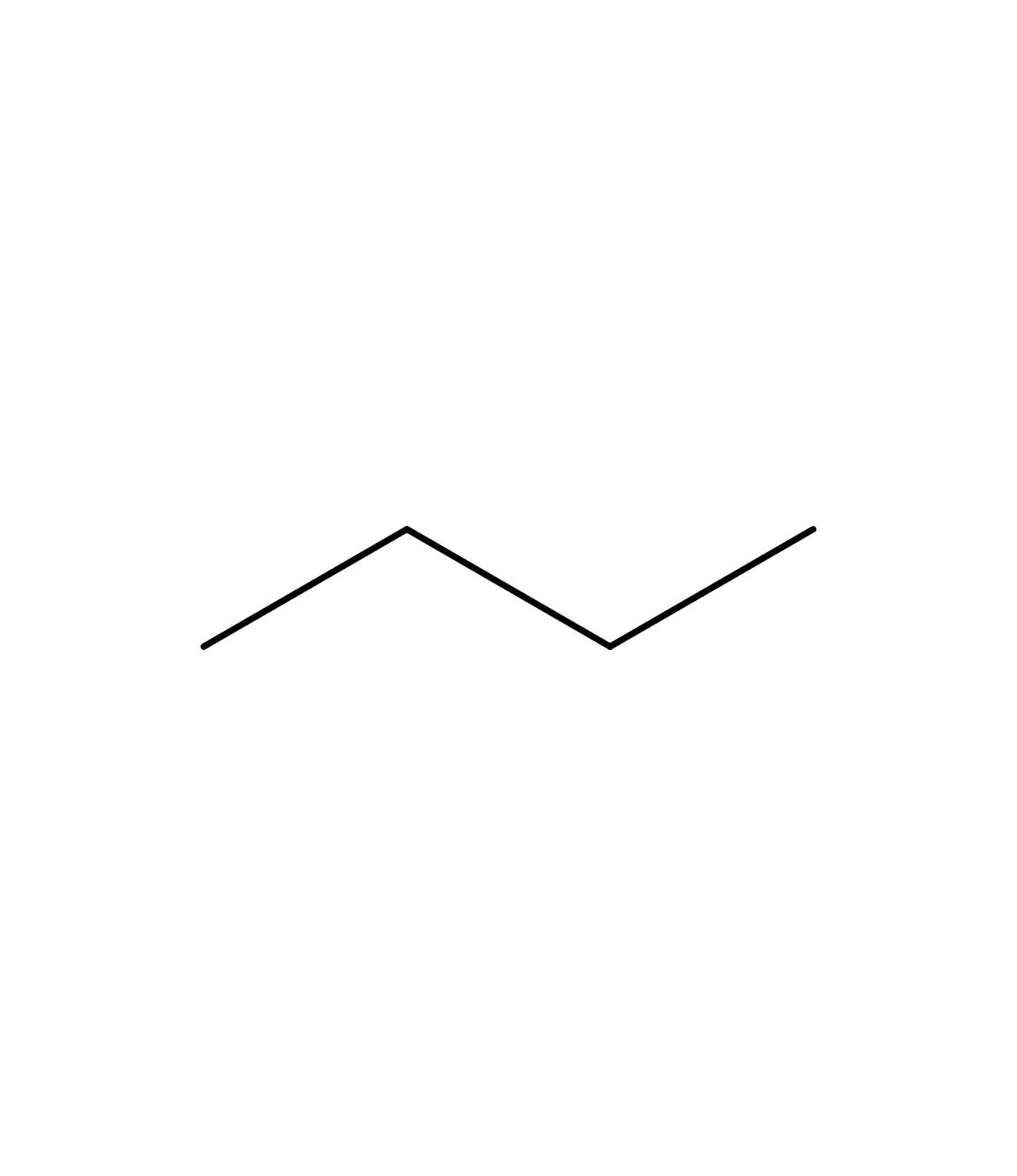 | CCCC |
| エタノール | 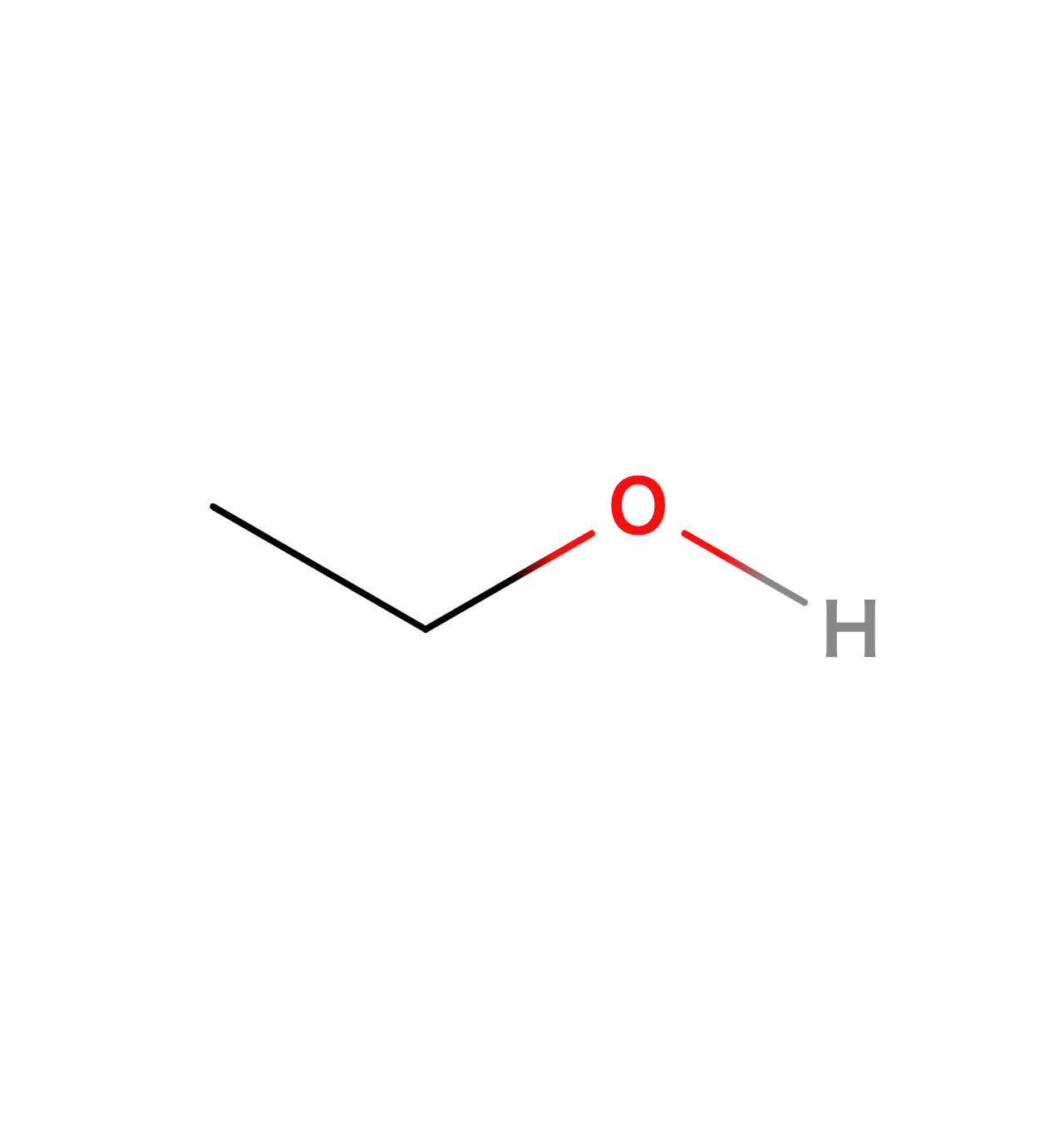 | CCO |
| エチルアミン | 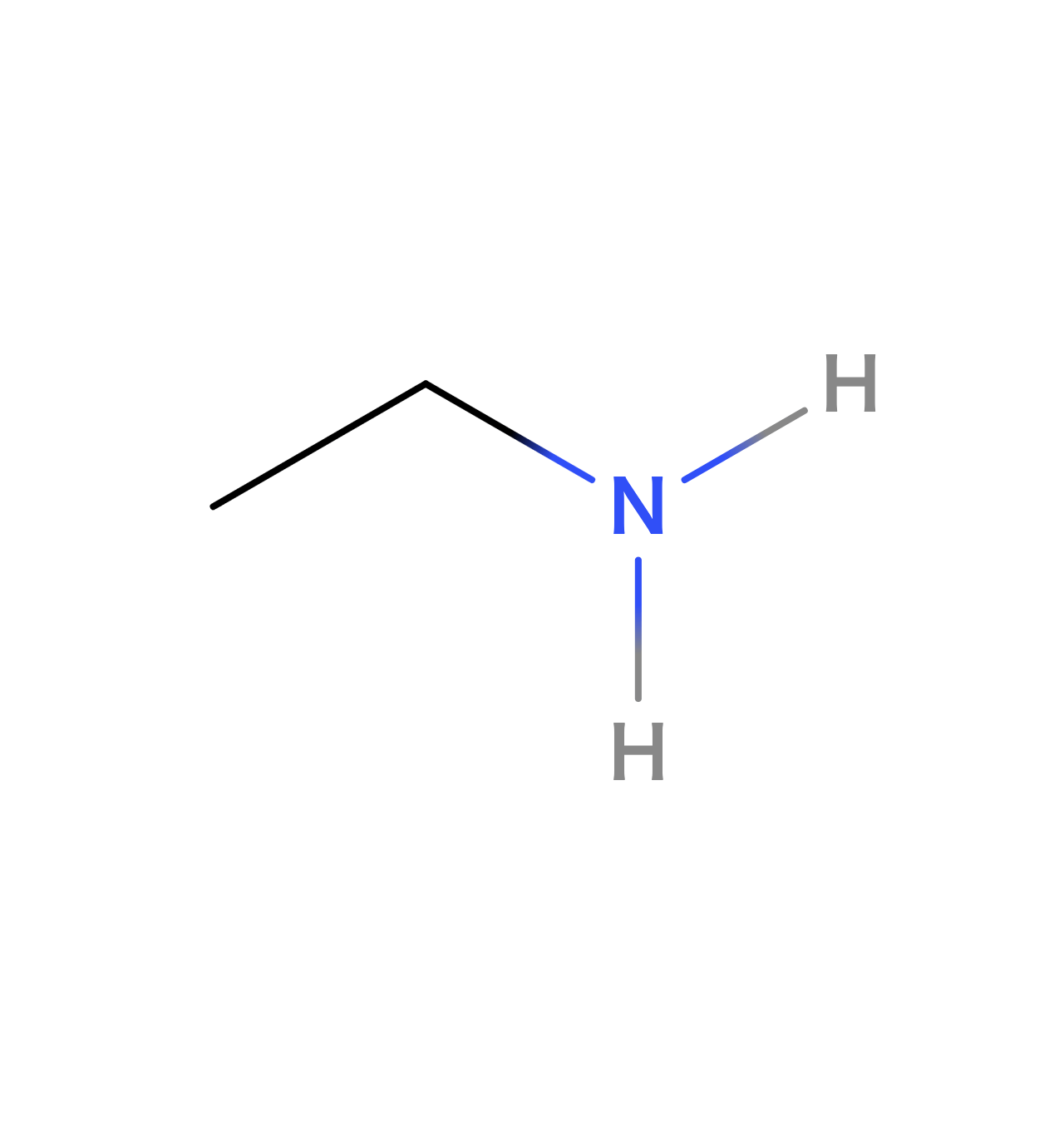 | CCN |
水素原子
SMILESにおいて水素原子は通常は省略されます。なので、先ほどご紹介したブタン、エタノール、エチルアミンでも水素は省略されていることがわかると思います。
しかし、水素原子を意図的に表記したい場合であれば[H]という形で表記できます。
結合
結合は単結合であれば省略されます。しかし、二重結合は「=」、三重結合は「#」で表記されるルールとなっております。以下に例として1-ブテンとアセチレンのSMILES表記をご紹介します。
| 化合物 | 構造式 | SMILES表記 |
|---|---|---|
| 1-ブテン | 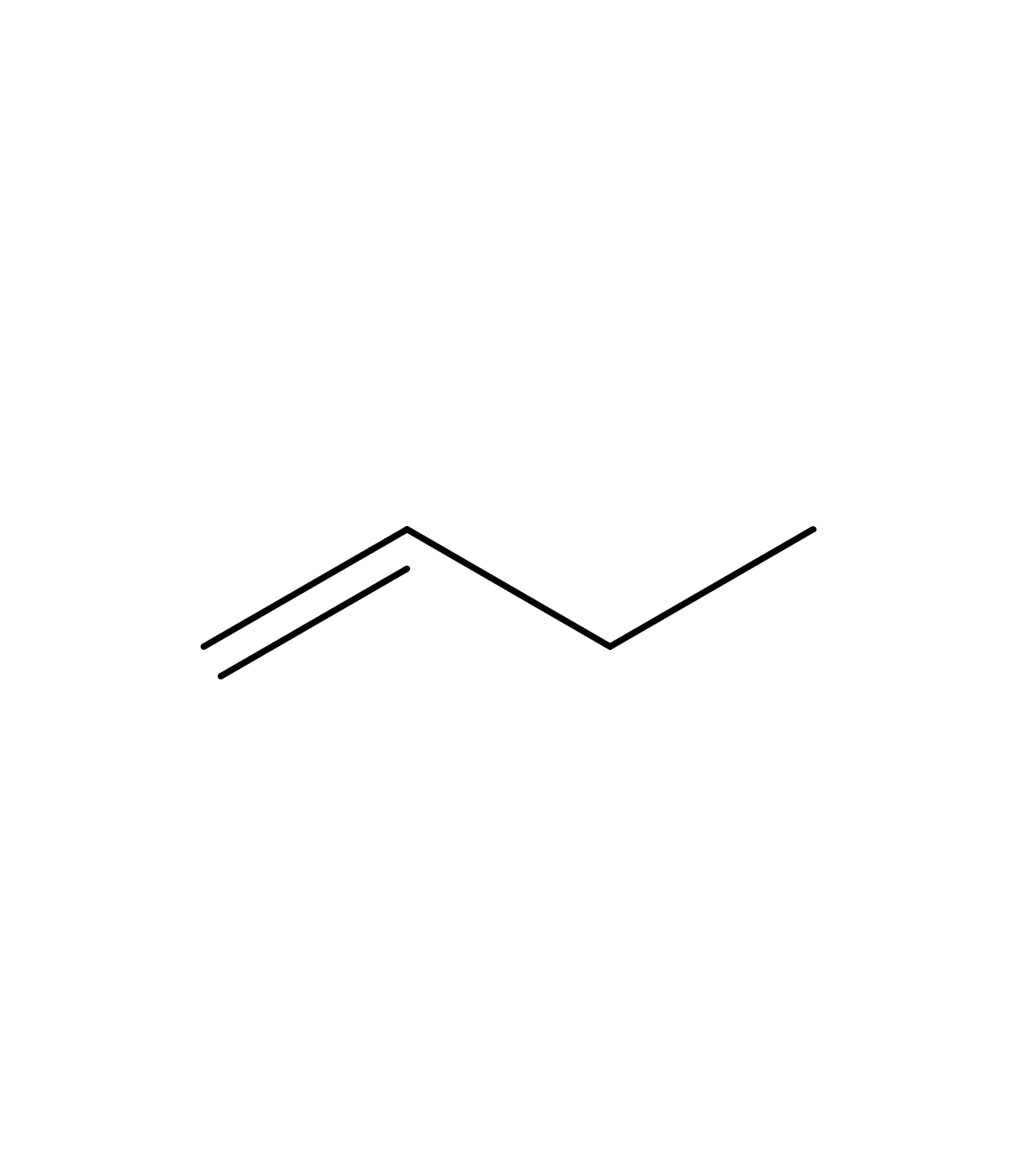 | CCC=C |
| アセチレン |  | C#C |
環構造
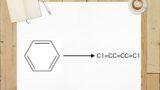
環構造は環の最初と最後に数字を記載して直鎖として表記します。例としてベンゼン、ピリジンをSMILES表記にしたものを示します。
| 化合物 | 構造式 | SMILES表記 |
|---|---|---|
| ベンゼン | 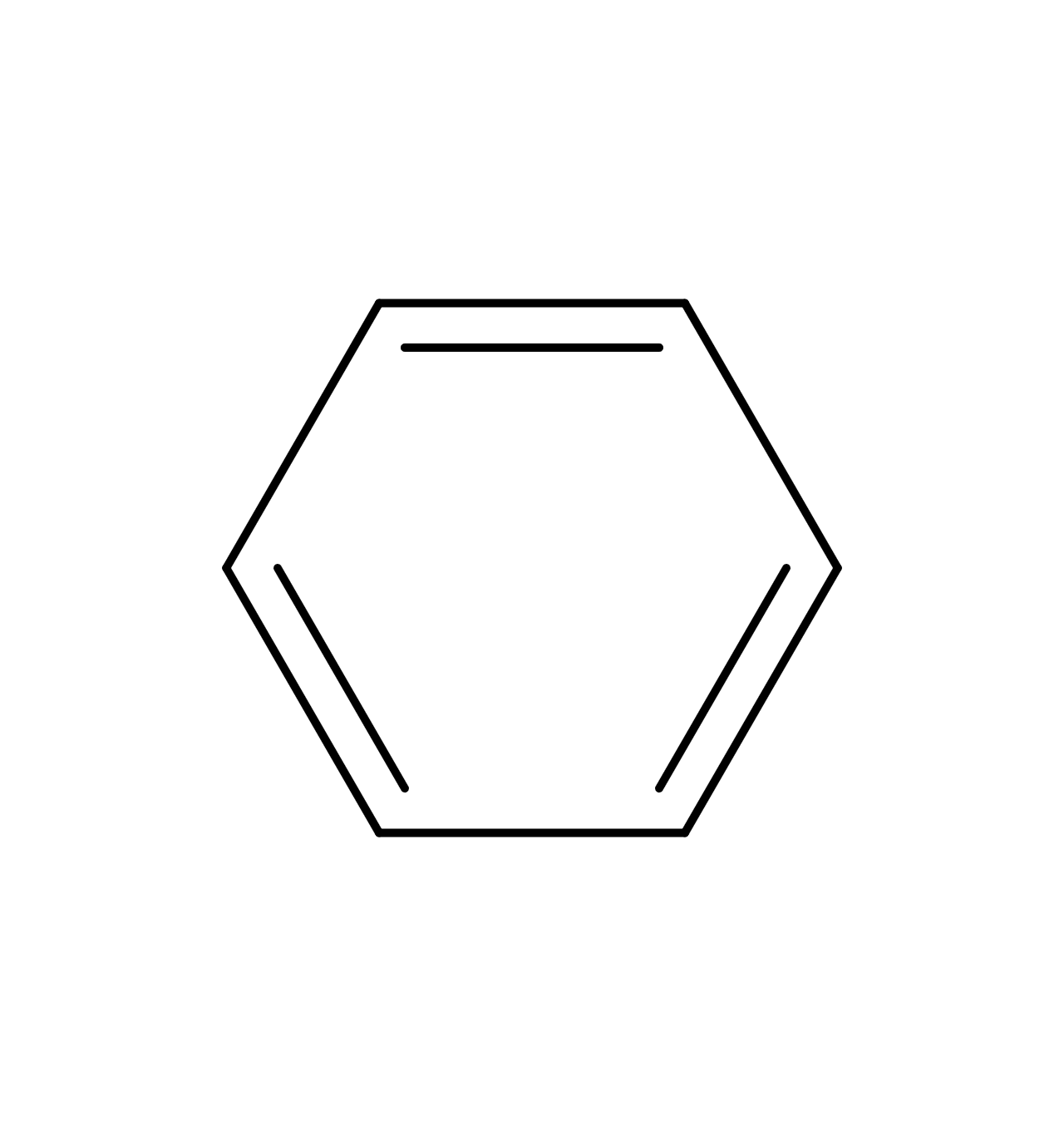 | C1=CC=CC=C1 |
| ピリジン | 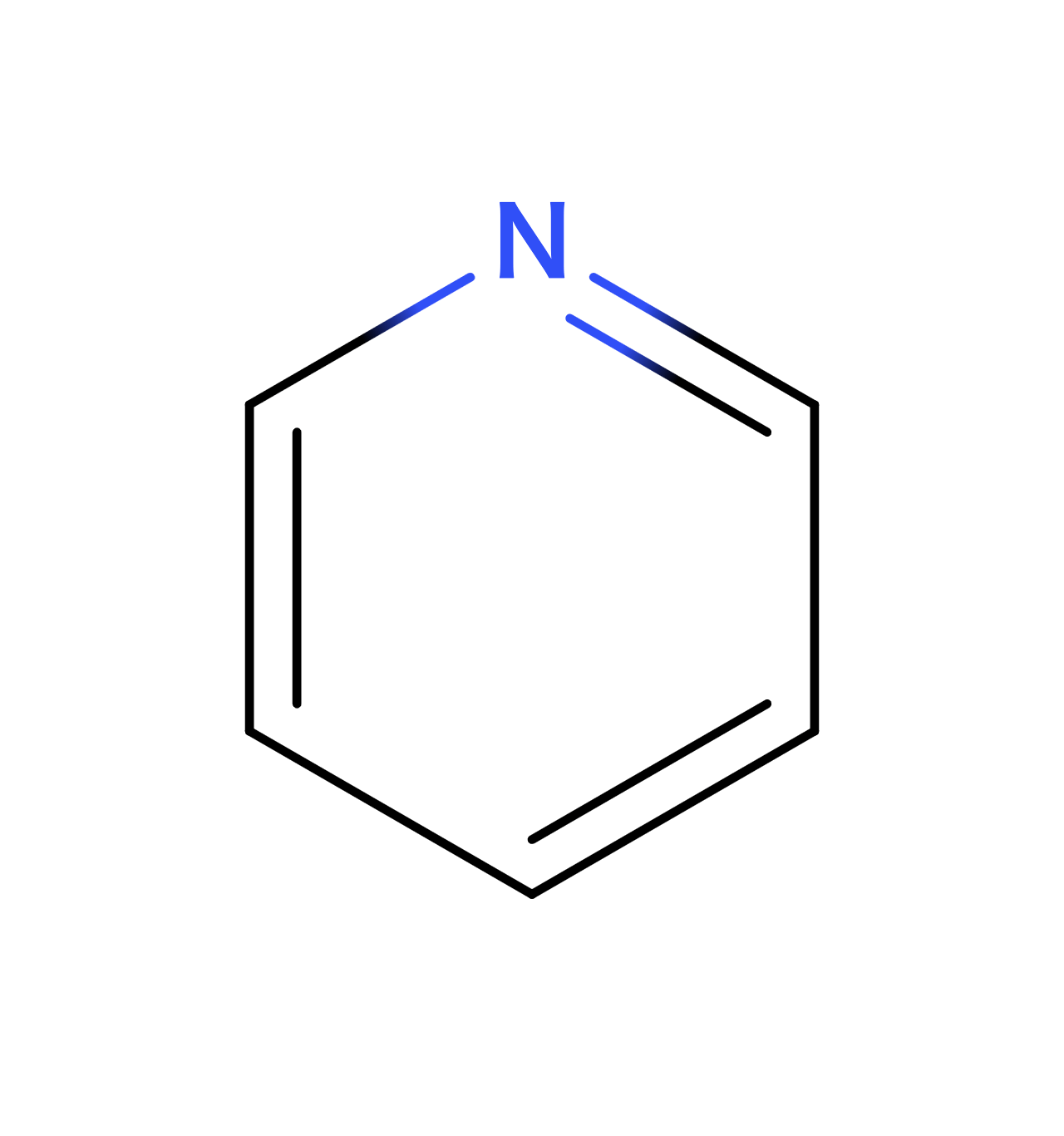 | C1=CC=NC=C1 |
分岐構造
分岐構造の場合は「()」で分岐の部分構造を括ることで表現しています。以下に2-メチルペンタン、2-ブタノールのSMILES表記を示します。
| 化合物 | 構造式 | SMILES表記 |
|---|---|---|
| 2-メチルペンタン | 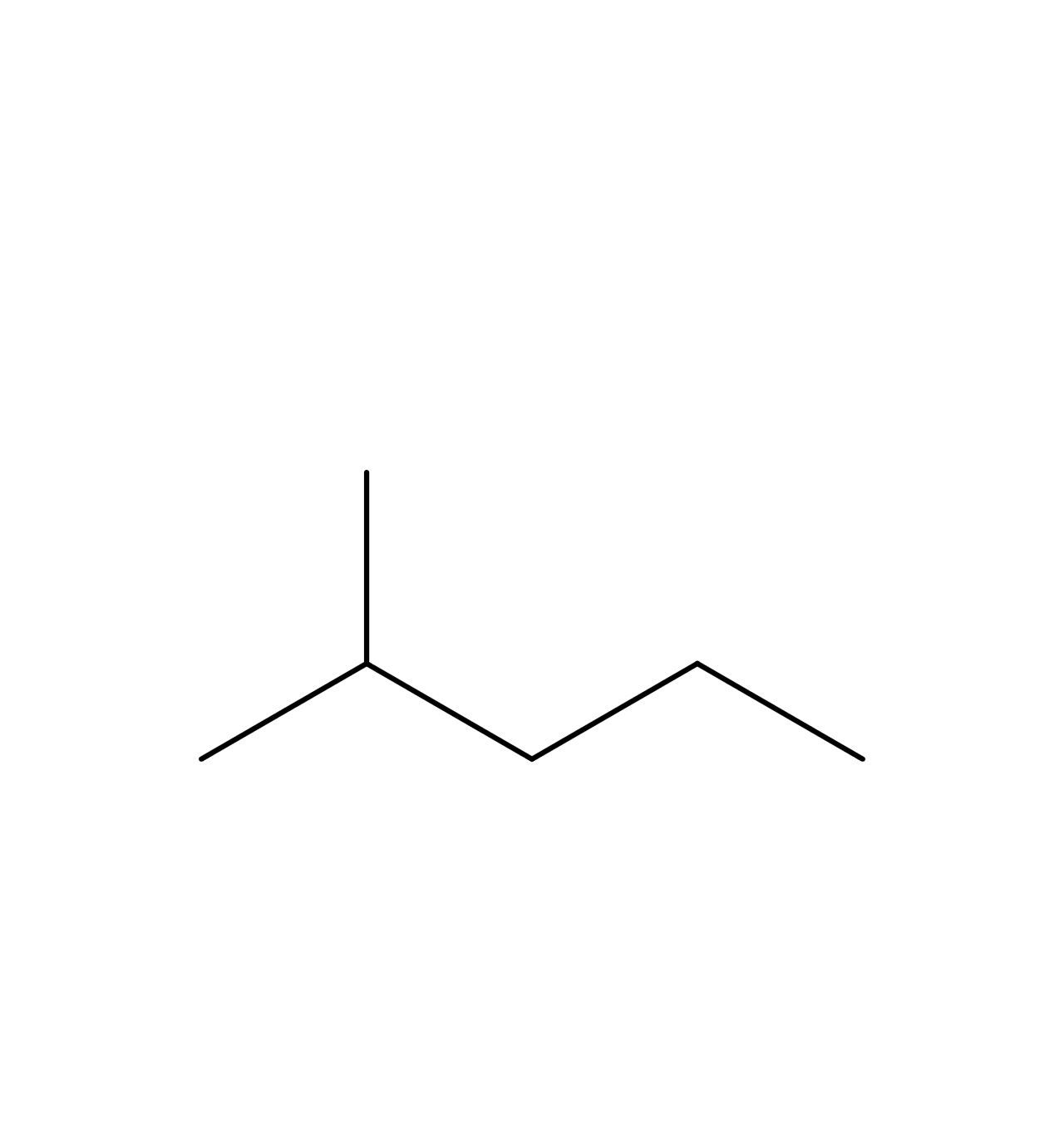 | CCCC(C)C |
| 2-ブタノール | 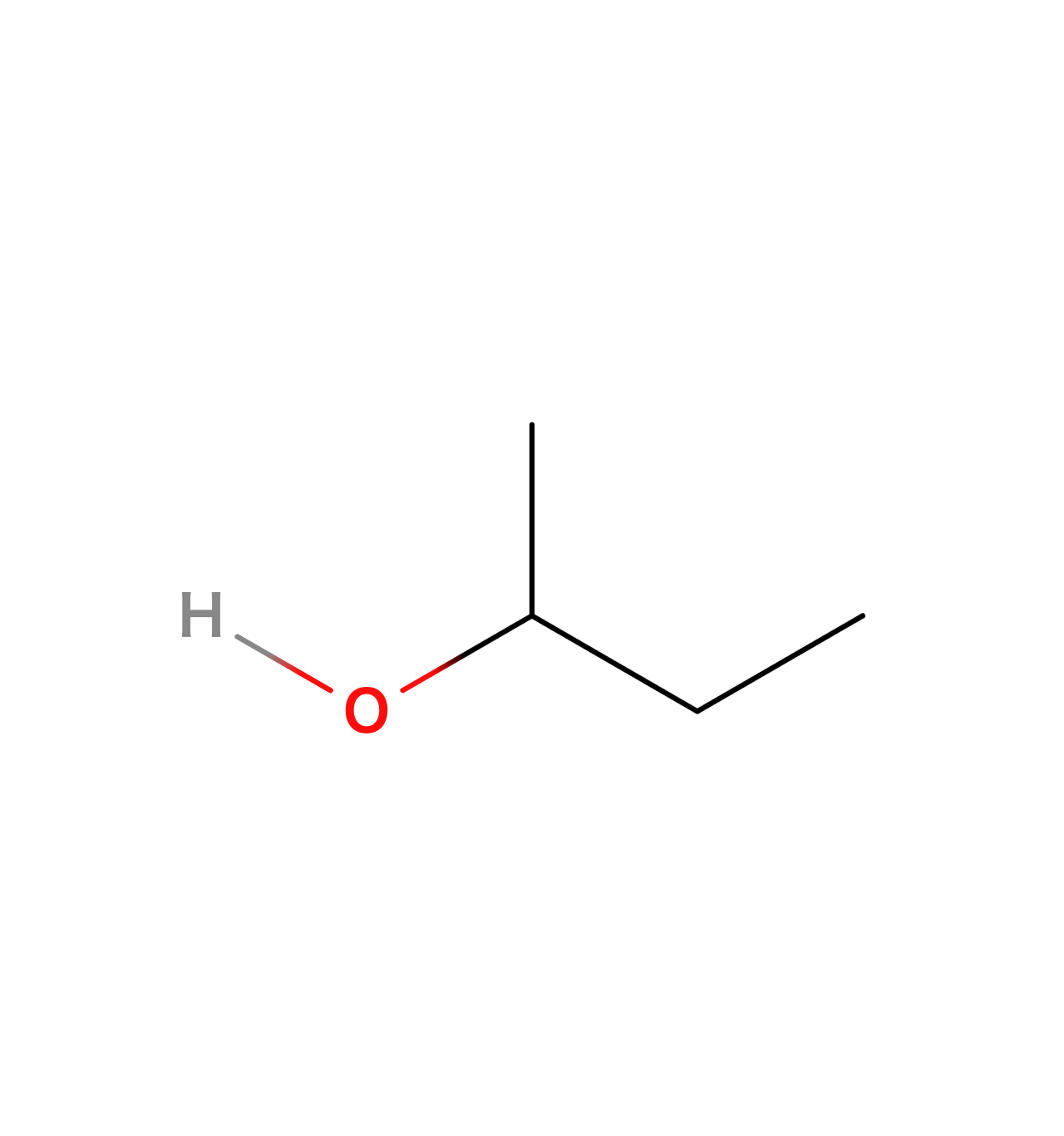 | CCC(C)O |
幾何異性体
cis-, trans-などの幾何異性体の場合は「/」や「\」を使用して表記します。以下に例としてtrans-2-ブテン、cis-2-ブテンのSMILES表記をご紹介します。
| 化合物 | 構造式 | SMILES表記 |
|---|---|---|
| trans-2-ブテン | 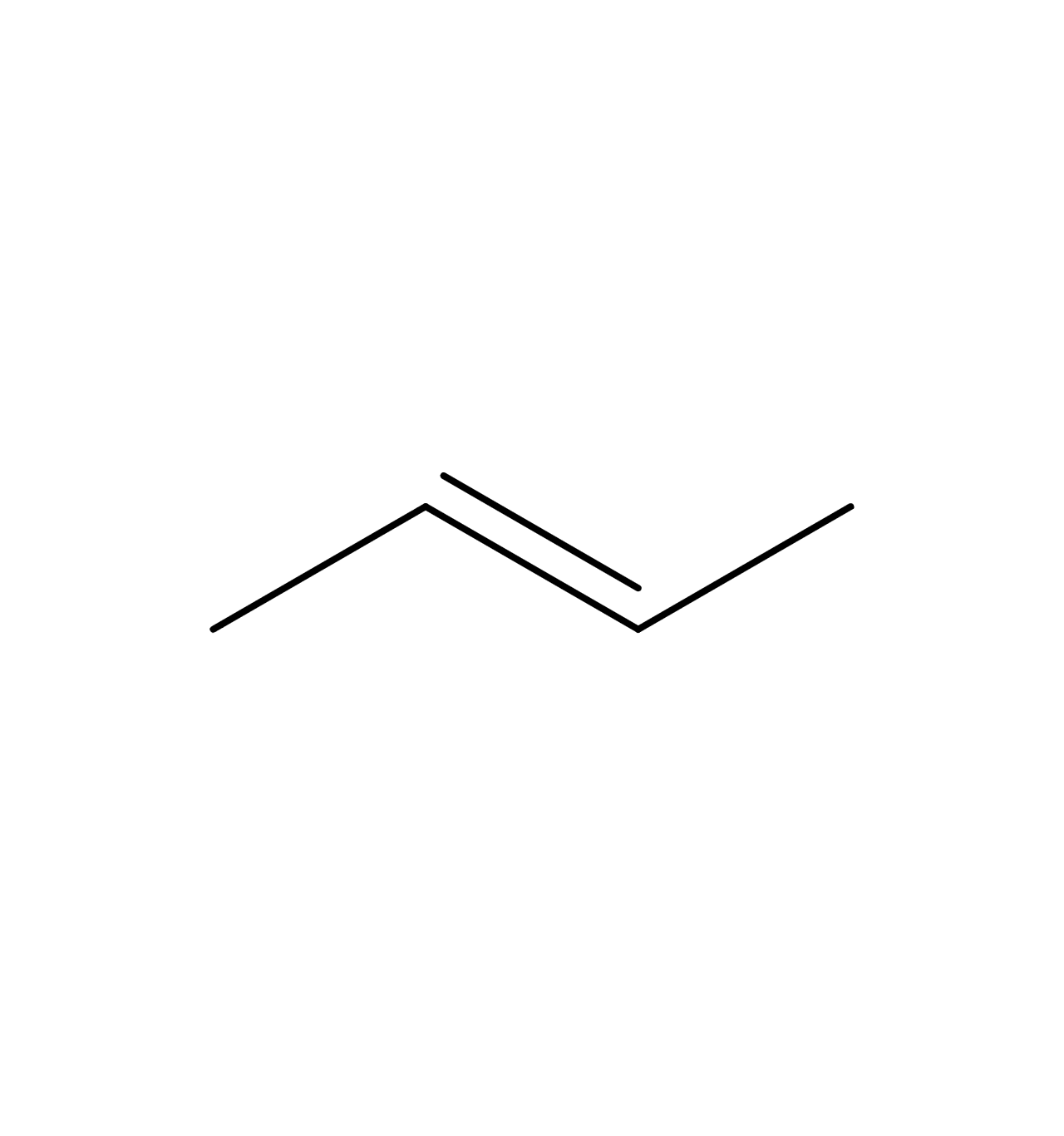 | C/C=C/C |
| cis-2-ブテン |  | C/C=C\C |
他にもまだありますが、よく使用するルールとしては今ご紹介したものになるかと思います。
SMILESの取得方法
最後にSMILESの取得方法について解説します。
この記事では以下の通り、大きく3つの方法についてご紹介します。
- MolView
- SMILES generator
- Pythonによる自動取得
それでは順番に解説していきます。
MolView
まずはMolViewを使用してSMILESを取得する方法についてです。
MolViewは、分子の3D構造を表示し、化学情報を可視化するための化学ソフトウェアプラットフォームでありwebサイトで利用することができます。
MolViewは無料で利用でき、分子モデリングや可視化に役立つツールで描画した化合物のSMILESを取得することができます。
SMILESの取得方法としてはまず、以下のURLからMolViewにアクセスしてください。
MolViewのリンク↓↓↓↓
https://molview.org/
次に下の図のようにSMILESを取得したい化合物を描画します。今回はサリチル酸を描画しました。
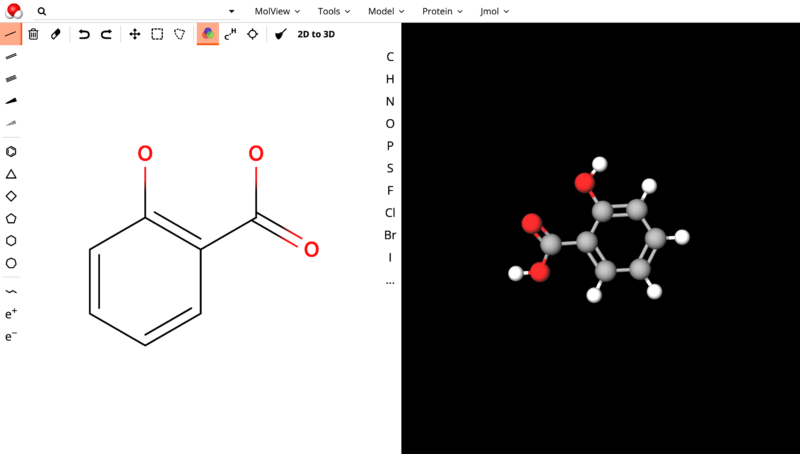
次に下の図の赤枠にあるTools内の「Information card」を選択します。
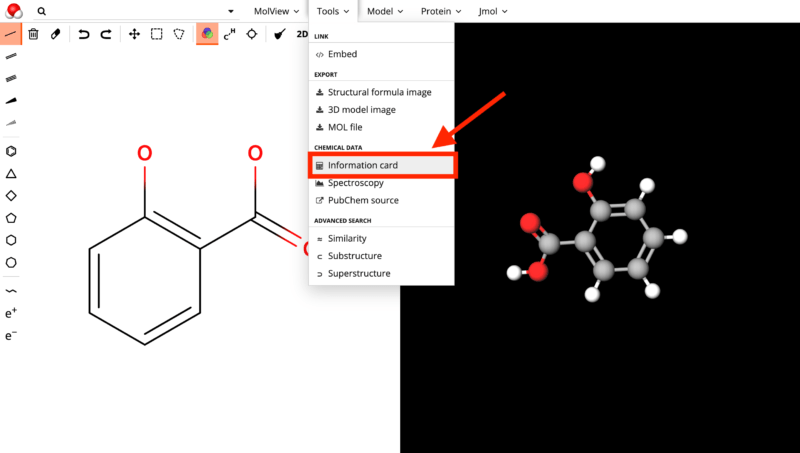
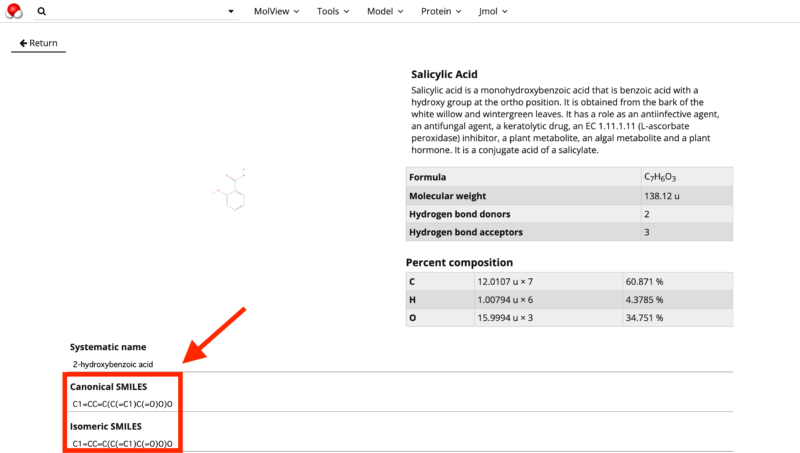
すると、下の赤枠内にあるようにcanonical SMILESとisomeric SMILESが取得できます。
canonical SMILESは正規表現されたSMILESであり、同じ分子であれば同じSMILESで表されます。
isomeric SMILESは立体的な構造も表現できるSMILESになります。どちらもSMILESに変わりありませんが、データ解析する際にはどちらかに統一しないと正しく分析できないことがあるので注意です。
SMILES generator
続いてはSMILES generatorを使用したSMILESの取得についてご紹介します。
SMILES generatorも無料で利用することができ、web上で利用することができます。
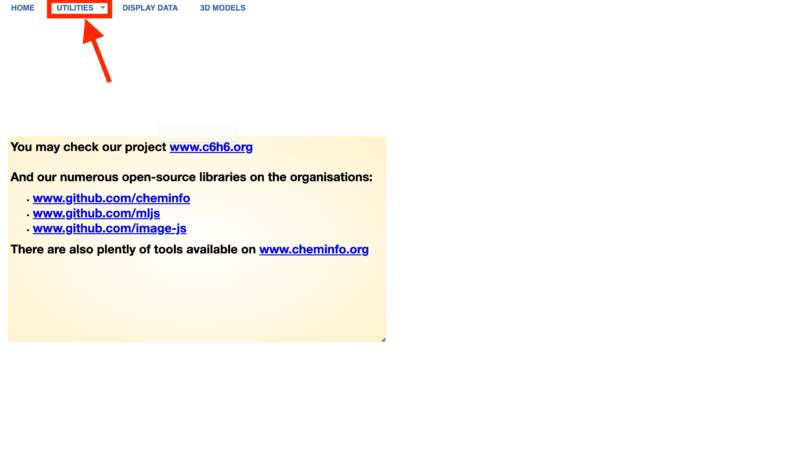
具体的なSMILESの取得方法ですが、まず下記URLからSMILES generatorにアクセスします。
SMILES generator↓↓↓↓
https://www.cheminfo.org/flavor/malaria/index.html
次に下図の赤枠のように「UTILITIES」を選択します。
次にUTILITIESの中の「SMILES generator」を選択します。
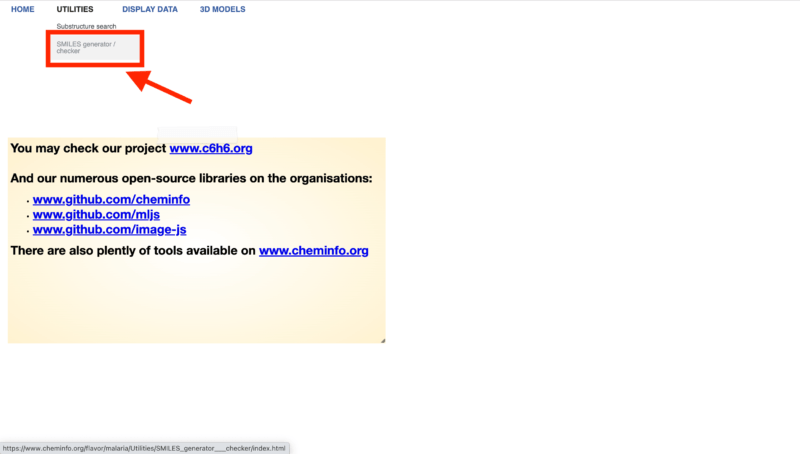
すると下の図の様に化合物が描画できるのでSMILESを取得したい化合物を描画しましょう。
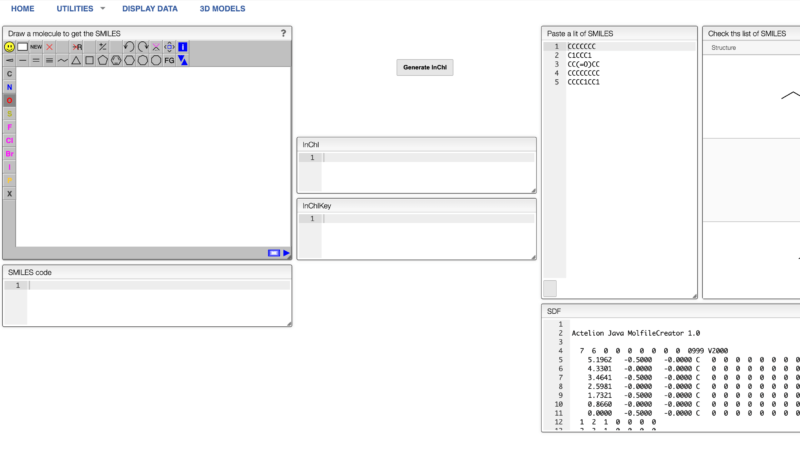
描画したら下の図の赤枠にあるニコちゃんマークをクリックします。
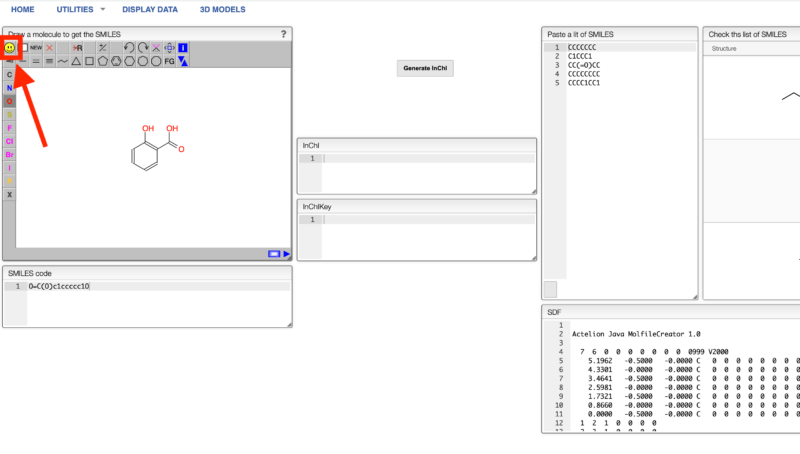
するとSMILESの式が取得できます。
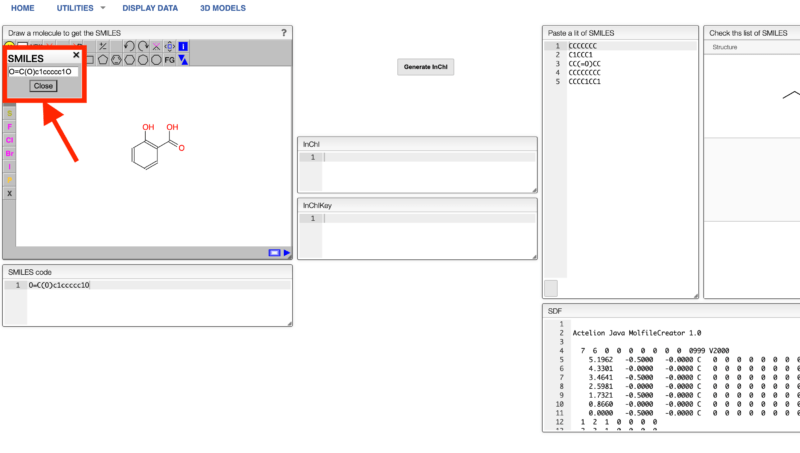
Pythonによる自動取得
最後にPythonによる自動取得についてご紹介します。
Pythonではpubchempyというライブラリを使用してPubChemから自動でSMILESを取得します。
今回は化合物のCAS No.からSMILESを自動取得するコードを作成しました。
以下が実際のコードになります。
import pubchempy as pcp
# CAS番号を指定
cas_number = "69-72-7" # ここにCAS番号を入力
# PubChemで化合物を検索
compounds = pcp.get_properties('CanonicalSMILES', cas_number, 'name', record_type='3d')
# SMILESを取得
if compounds:
compound = compounds[0] # 一番上の結果を取得
smiles = compound['CanonicalSMILES']
print(f"CAS番号 {cas_number} のSMILESは: {smiles}")
else:
print(f"CAS番号 {cas_number} に対する結果が見つかりませんでした。")
#出力結果
#CAS番号 69-72-7 のSMILESは: C1=CC=C(C(=C1)C(=O)O)O
今回はサリチル酸のCAS No,からSMILESを自動取得しましたが、うまく取得できている様ですね。
SMILESの活用例
SMILESは単なる構造表記にとどまらず、さまざまな化学・情報処理の分野で活用されています。以下に活用方法として「分子の類似性評価」と「機械学習モデルによる物性予測」の2つを紹介します。
分子の類似性評価(Tanimoto係数)
SMILES表記を用いて分子同士の類似度を数値化することができます。よく使われる手法が Tanimoto係数 です。
Tanimoto係数やその他化合物の類似度の評価手法については過去にまとめた記事があるので興味のある方はご参照ください。
流れとしては以下の通りです。
- SMILESからフィンガープリント(0 or 1のバイナリ特徴ベクトル)を生成
- 2つのベクトル間のTanimoto係数を計算
フィンガープリントは、分子の構造や性質をバイナリビット(0または1)や数値の形式で表現するものであり、分子構造の特定のパターンや特性を分析することができます。
フィンガープリントについては過去の記事でまとめていますのでご参考にしていただければと思います。
化合物の類似度計算については様々な応用例がありますが、以下のように創薬分野や化合物データの解析にも使用されたりします。
- 創薬分野における類似化合物の検索(ドラッグリポジショニング)
→既存薬の別疾患への活用
- 化合物ライブラリのクラスタリング
→化合物DBのクラスタリングによる分類
では実際にPythonでSMILESからtanimoto係数の計算を実行してみます。
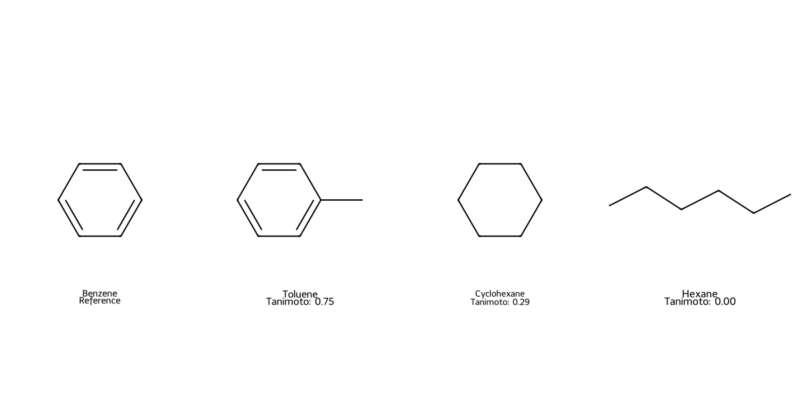
今回はベンゼンに対してトルエン、シクロヘキサン、n-ヘキサンのTanimoto係数を計算してみました。
実行コードは以下の通りです。
from rdkit import Chem
from rdkit.Chem import MACCSkeys, Draw
from rdkit.DataStructs import FingerprintSimilarity
# ベンゼン、トルエン、シクロヘキサン、ヘキサンのSMILES表現
smiles = {
'Benzene': 'c1ccccc1',
'Toluene': 'Cc1ccccc1',
'Cyclohexane': 'C1CCCCC1',
'Hexane': 'CCCCCC'
}
# 化合物の名前と分子を格納するリスト
molecules = [(name, Chem.MolFromSmiles(smile)) for name, smile in smiles.items()]
# ベンゼンの分子オブジェクトとMACCSフィンガープリント
benzene_fp = MACCSkeys.GenMACCSKeys(molecules[0][1])
# Tanimoto係数を計算する関数
def calculate_tanimoto(smiles, reference_fp):
mol = Chem.MolFromSmiles(smiles)
fp = MACCSkeys.GenMACCSKeys(mol)
return FingerprintSimilarity(reference_fp, fp)
# 各化合物のTanimoto係数を計算し、タイトルに追加
for i, (name, mol) in enumerate(molecules):
if name != 'Benzene':
similarity = calculate_tanimoto(smiles[name], benzene_fp)
molecules[i] = (f'{name}\nTanimoto: {similarity:.2f}', mol)
else:
molecules[i] = (f'{name}\nReference', mol)
# 分子を描画し、タイトルを追加
img = Draw.MolsToGridImage([mol for name, mol in molecules],
legends=[name for name, mol in molecules],
molsPerRow=4,
subImgSize=(300, 300))結果を出力すると以下のように計算されていました。トルエン→シクロヘキサン→n-ヘキサンの順でTanimoto係数が大きく、類似していることになります。n-ヘキサンは0なので、全く似ていないという結果になっていますね。
機械学習モデルによる物性予測
SMILESを用いると、回帰や分類モデルを構築することができます。
RDKitなどのPythonライブラリを使えば、SMILESから数百種類の分子記述子を自動で抽出可能です。
流れとしては以下の通りです。
- SMILESから分子記述子(Mordred記述子など)を生成
- 予測したい物性(融点、物性、毒性など)に対して分子記述子を説明変数とした機械学習モデルを構築
- 予測したい化合物の分子記述子から目的物性を予測
Mordred記述子については過去の記事でまとめていますので、ご参考にしていただければと思います。
使用した機械学習モデルはランダムフォレスト回帰です。ランダムフォレストについての詳細は過去に記事をまとめていますので、ご興味あれば参照いただければと思います。

では実際にSMILESを用いた化合物の物性予測をPythonを使用して実行していきます。
今回はSMILESからMordred記述子に変換してxlogp(水溶性か脂溶性かを測る指標)を予測していきます。なお、学習データ1600点に対してテストデータ400点を予測させています。
実行したPythonコードは以下の通りとなります。
import pandas as pd
from rdkit import Chem
from mordred import Calculator, descriptors
from sklearn.ensemble import RandomForestRegressor
from sklearn.model_selection import train_test_split
from sklearn.metrics import mean_squared_error, r2_score
import numpy as np
import matplotlib.pyplot as plt #
# === 1. データ読み込みと前処理 ===
df = pd.read_csv("dataset.csv", index_col=0, header=0)
df = df.dropna(subset=["xlogp"]).copy()
df["Mol"] = df["canonicalsmiles"].apply(Chem.MolFromSmiles)
# === 2. Mordred 記述子の計算 ===
calc = Calculator(descriptors, ignore_3D=True)
desc_df = calc.pandas(df["Mol"])
# === 3. xlogp と記述子を結合 ===
df_full = pd.concat([df[["xlogp"]], desc_df], axis=1)
# === 4. 数値型のみを抽出し、NaN・infを除去 ===
df_numeric = df_full.select_dtypes(include=[np.number])
df_numeric["xlogp"] = df_full["xlogp"]
df_numeric = df_numeric.replace([np.inf, -np.inf], np.nan).dropna()
# === 5. 特徴量と目的変数に分離 ===
X = df_numeric.drop(columns=["xlogp"])
y = df_numeric["xlogp"]
# === 6. モデル構築・評価 ===
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
model = RandomForestRegressor(n_estimators=100, random_state=42)
model.fit(X_train, y_train)
y_pred = model.predict(X_test)
# === 7. 評価指標 ===
print("MSE:", mean_squared_error(y_test, y_pred))
print("R² :", r2_score(y_test, y_pred))
# === 8. 可視化:実測値 vs 予測値 ===
plt.figure(figsize=(8, 6))
plt.scatter(y_test, y_pred, alpha=0.7, edgecolors='k')
plt.plot([y_test.min(), y_test.max()], [y_test.min(), y_test.max()], 'r--') # 理想線
plt.xlabel("Actual xlogP")
plt.ylabel("Predicted xlogP")
plt.title("Actual vs Predicted xlogP")
plt.grid(True)
plt.tight_layout()
plt.show()
#出力結果
#MSE: 0.53
#R² : 0.93
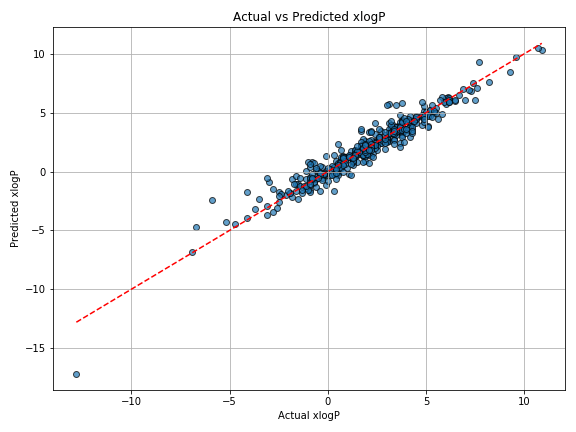
出力結果を見ると $R^2$ 値は0.93程度と良好なモデルを構築できていると思われます。
テストデータの予測値に対する実測値は以下の図の通りとなります。赤い点線上にデータが近いほど予測値と実測値が一致していることを表しており、このグラフを見ると非常に予測精度が良好であることがわかりますね。
以上がSMILESの活用例になります。
オススメの書籍
最後に、SMILESの理解をさらに深めたい方、SMILESを活用したデータ分析や機械学習に挑戦したい方に向けて、ぜひ手に取っていただきたい一冊をご紹介します。

ご紹介する『化学のためのPythonによるデータ解析・機械学習入門』では、SMILESをはじめとする化学構造のテキスト表記方法から、それらをPythonで数値化する方法まで、実践的なサンプルコード付きで丁寧に解説されています。
- SMILES記法の基本から応用まで丁寧に解説
- Pythonコード付きで即実践できる
- 化学構造のデータ化 → 機械学習まで一連の流れがわかる
- 実例豊富でイメージしやすい
- 数式少なめ、やさしい解説なので、初学者にも安心
上記の通り、本書籍の特徴として実際のデータセットを使ったデータ分析・機械学習の事例も豊富に紹介されており、単なる理論にとどまらず、研究や開発現場ですぐに応用できる実践力が身につきます。
難易度についても、タイトル通り入門書にふさわしく、高度な数学的知識は不要。大学の基礎レベルの知識があれば、スムーズに理解できる構成になっていますので、初めてこの分野に取り組む方でも安心です。
「SMILESを本当に使いこなせるようになりたい」、「化学×データサイエンスの第一歩を踏み出したい」という方に、強くおすすめできる一冊です!
終わりに
以上がSMILES表記についての解説になります。機械学習などでは化学構造をSMILES表記に変換して構造情報などを学習させることがあります。また、データベース化する際にSMILESの情報を入力しておけば類似構造などで構造を検索することができますね。
ちなみにSMILES表記はchemdrawやmolviewなどの描画ソフトで構造を描画した際に自動でSMILES表記にしてくれる機能が備わっていますので、実際にSMILES表記を使用する際は自分で表記を考えることはほぼありません。一つずつ表記を考えていたら間違いもありますし、膨大な時間もかかりますからね…
ぜひ、構造情報をデータベース化したいなどありましたらSMILES表記をご使用ください!
ちなみに今回とは逆にSMILESから化学構造を描画する方法については以下の記事にまとめています。